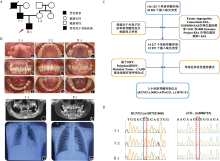
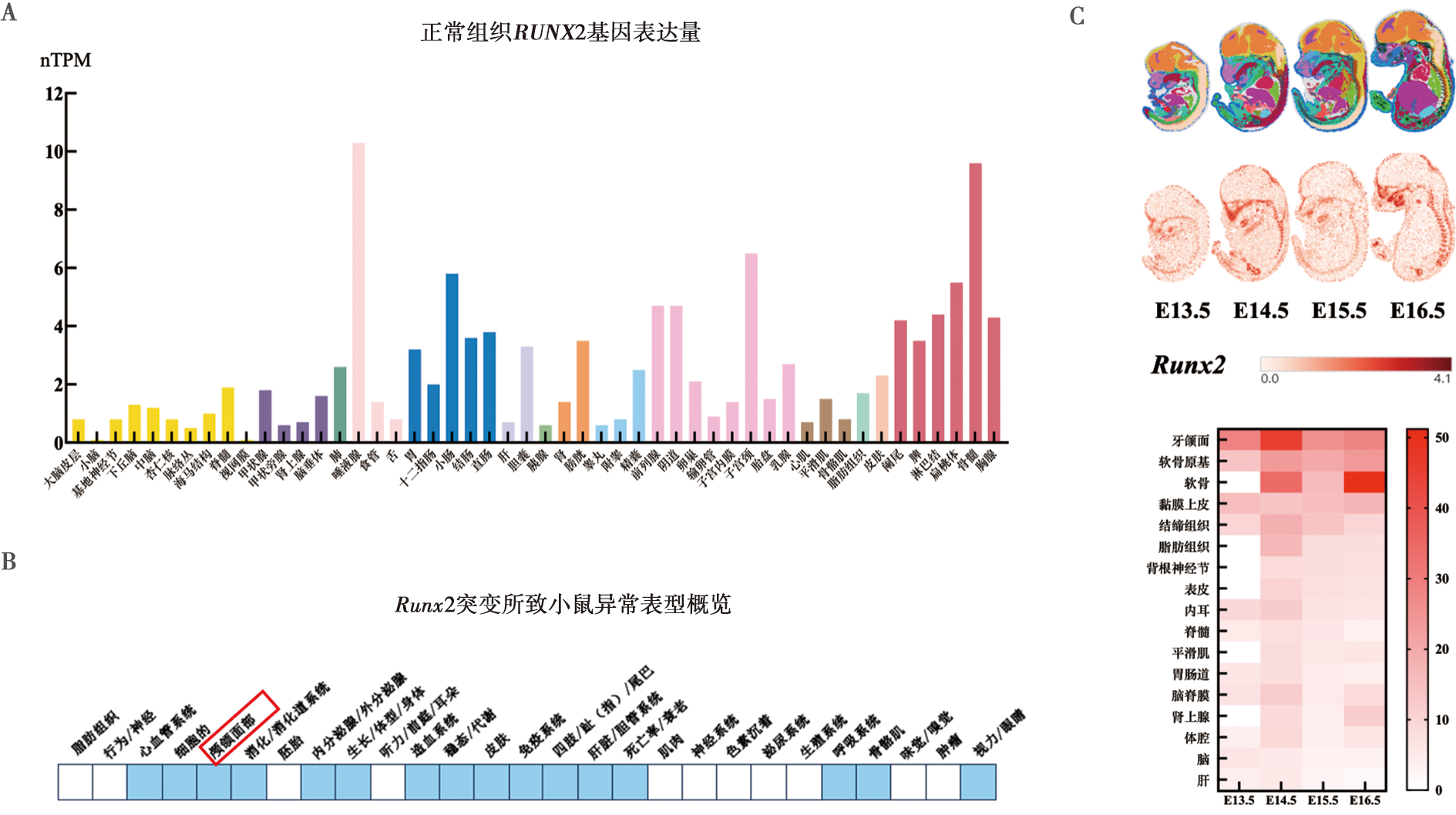
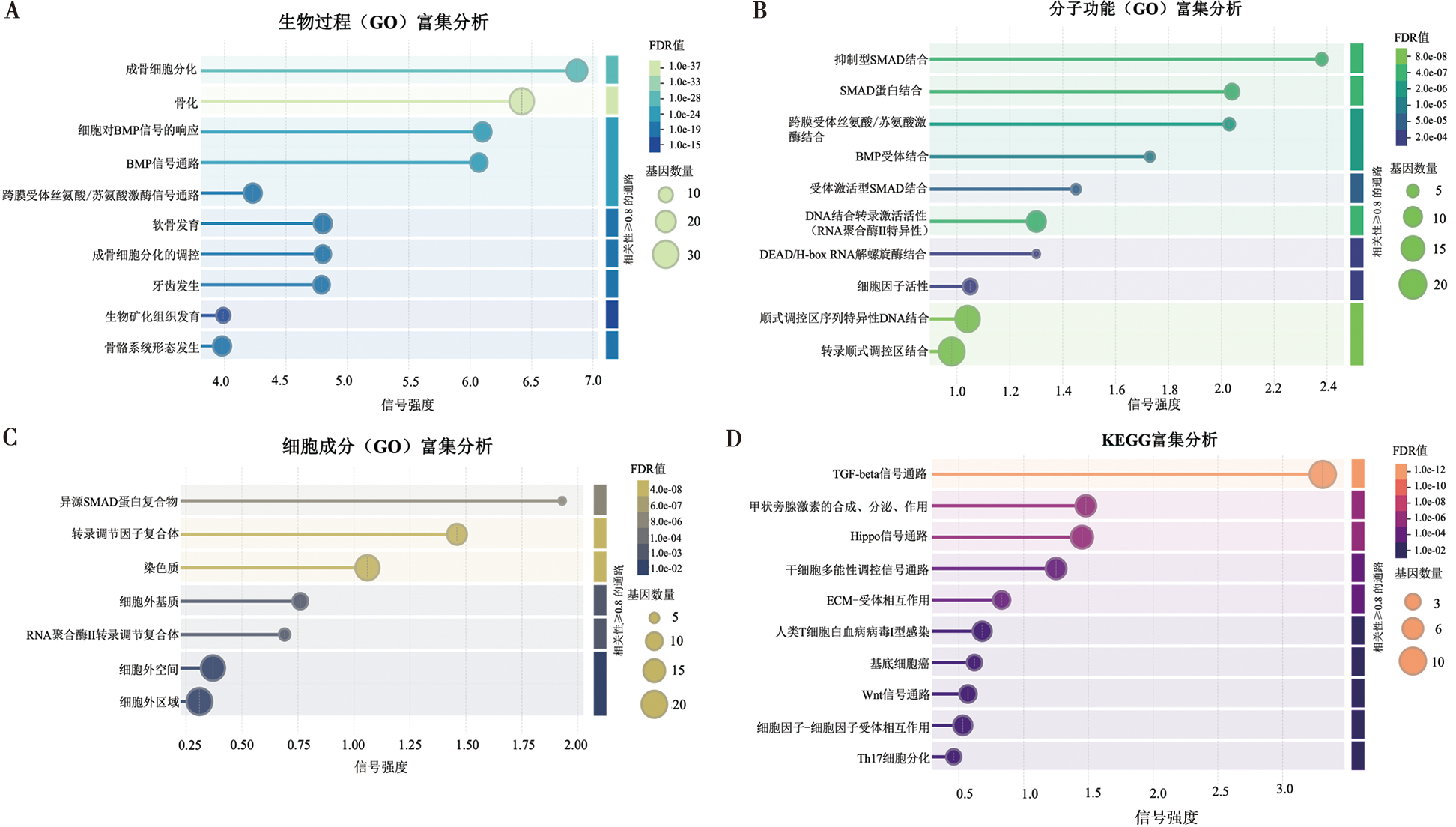
| [1] |
张鹏, 何平华, 徐佩琼, 等. 颅骨锁骨发育不全1例及基因检测分析[J]. 华西口腔医学杂志, 2022, 40(3): 360-364.
|
| [2] |
Pan CY, Tseng YC, Lan TH, et al. Craniofacial features of cleidocranial dysplasia[J]. J Dent Sci, 2017, 12(4): 313-318.
doi: 10.1016/j.jds.2017.07.002
|
| [3] |
潘育桦, 谢远雯, 王贺, 等. 颅骨锁骨发育不全RUNX2基因错义突变1例[J]. 临床口腔医学杂志, 2021, 37(8): 450, 475.
|
| [4] |
姜涛, 蒋序, 张运奎. 一颅锁骨发育不全综合征家族的RUNX2基因分析研究[J]. 华西口腔医学杂志, 2013, 31(5): 522-525.
|
| [5] |
梁秋梅, 韩路军, 刘红艳. 颅锁骨发育不全综合征临床、影像表现并文献复习[J]. 罕少疾病杂志, 2021, 28(1): 55-57.
|
| [6] |
Mundlos S, Otto F, Mundlos C, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia[J]. Cell, 1997, 89(5): 773-779.
doi: 10.1016/s0092-8674(00)80260-3
pmid: 9182765
|
| [7] |
付东杰, 佘文婷, 李俊, 等. 颅骨锁骨发育不全(CCD)患者RUNX2基因突变的研究[J]. 临床口腔医学杂志, 2018, 34(10): 582-585.
|
| [8] |
赵志河. 口腔正畸学[M]. 7版. 北京: 人民卫生出版社, 2020.
|
| [9] |
Otto F, Kanegane H, Mundlos S. Mutations in the RUNX2 gene in patients with cleidocranial dysplasia[J]. Hum Mutat, 2002, 19(3): 209-216.
doi: 10.1002/humu.10043
pmid: 11857736
|
| [10] |
Cooper SC, Flaitz CM, Johnston DA, et al. A natural history of cleidocranial dysplasia[J]. Am J Med Genet, 2001, 104(1): 1-6.
doi: 10.1002/(ISSN)1096-8628
|
| [11] |
Vimalraj S, Arumugam B, Miranda PJ, et al. Runx2: Structure, function, and phosphorylation in osteoblast differentiation[J]. Int J Biol Macromol, 2015, 78: 202-208.
doi: 10.1016/j.ijbiomac.2015.04.008
pmid: 25881954
|
| [12] |
Chen YB, Zhao XY, Wu H. Transcriptional programming in arteriosclerotic disease: A multifaceted function of the Runx2 (runt-related transcription factor 2)[J]. Arterioscler Thromb Vasc Biol, 2021, 41(1): 20-34.
doi: 10.1161/ATVBAHA.120.313791
pmid: 33115268
|
| [13] |
Yi HY, He YH, Zhu QH, et al. RUNX proteins as epigenetic modulators in cancer[J]. Cells, 2022, 11(22): 3687.
doi: 10.3390/cells11223687
|
| [14] |
Xiong Y, Zhou Y. From molecular mechanisms to therapeutic targets: Research progress of RUNX2 in cancer[J]. Adv Clin Med, 2025, 15(3): 1572-1579.
doi: 10.12677/acm.2025.153778
|
| [15] |
张丛, 杜娟, 姜艳, 等. 颅骨锁骨发育不全综合征两例及基因突变分析[J]. 中华骨质疏松和骨矿盐疾病杂志, 2017, 10(3): 239-245.
|
| [16] |
Ott CE, Leschik G, Trotier F, et al. Deletions of the RUNX2 gene are present in about 10% of individuals with cleidocranial dysplasia[J]. Hum Mutat, 2010, 31(8): E1587-E1593.
|
| [17] |
Thaweesapphithak S, Termteerapornpimol K, Wongsirisuwan S, et al. The impact of RUNX2 gene variants on cleidocranial dysplasia phenotype: A systematic review[J]. J Transl Med, 2024, 22(1): 1099.
doi: 10.1186/s12967-024-05904-2
pmid: 39627759
|
| [18] |
Zhou G, Chen Y, Zhou L, et al. CBFA1 mutation analysis and functional correlation with phenotypic variability in cleidocranial dysplasia[J]. Hum Mol Genet, 1999, 8(12): 2311-2316.
doi: 10.1093/hmg/8.12.2311
pmid: 10545612
|
| [19] |
Yang LY, Lu GQ, Shen WJ, et al. Whole-exome sequencing of a novel initiation Codon mutation in RUNX2 in a Chinese family with cleidocranial dysplasia[J]. Medicine (Baltimore), 2021, 100(45): e27746.
doi: 10.1097/MD.0000000000027746
|
| [20] |
Zhao S, Wang T, Yang H, et al. Neonatal familiar cleidocranial dysplasia: A case report[J]. Am J Case Rep, 2025, 26: e946322.
|
| [21] |
Yoshida T, Kanegane H, Osato M, et al. Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations[J]. Am J Hum Genet, 2002, 71(4): 724-738.
doi: 10.1086/342717
pmid: 12196916
|
| [22] |
Ogawa S, Higuchi S, Yoshimoto Y, et al. Functional impact of pathogenic runt domain mutations in Runx2 in vivo: Insights into the skeletal and dental anomalies of cleidocranial dysplasia[J]. bioRxiv, 2025: 2025.06.18.660258.
|
| [23] |
Jiang Q, Qin X, Nagano K, et al. Different requirements of CBFB and RUNX2 in skeletal development among calvaria, limbs, vertebrae and ribs[J]. Int J Mol Sci, 2022, 23(21): 13299.
doi: 10.3390/ijms232113299
|
| [24] |
Komori T. Whole aspect of Runx2 functions in skeletal develop-ment[J]. Int J Mol Sci, 2022, 23(10): 5776.
doi: 10.3390/ijms23105776
|
| [25] |
Chan WCW, Tan ZJ, To MKT, et al. Regulation and role of transcription factors in osteogenesis[J]. Int J Mol Sci, 2021, 22(11): 5445.
doi: 10.3390/ijms22115445
|
| [26] |
Guasto A, Cormier-Daire V. Signaling pathways in bone development and their related skeletal dysplasia[J]. Int J Mol Sci, 2021, 22(9): 4321.
doi: 10.3390/ijms22094321
|
| [27] |
Wu MR, Chen GQ, Li YP. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease[J]. Bone Res, 2016, 4: 16009.
doi: 10.1038/boneres.2016.9
pmid: 27563484
|
| [28] |
Zhu SY, Chen W, Masson A, et al. Cell signaling and transcriptional regulation of osteoblast lineage commitment, differentiation, bone formation, and homeostasis[J]. Cell Discov, 2024, 10(1): 71.
doi: 10.1038/s41421-024-00689-6
pmid: 38956429
|
| [29] |
Sun K, Guo JC, Guo Z, et al. The roles of the Hippo-YAP signalling pathway in cartilage and osteoarthritis[J]. Ageing Res Rev, 2023, 90: 102015.
doi: 10.1016/j.arr.2023.102015
|

 ), 潘永初1,2,3(
), 潘永初1,2,3( 苏公网安备32010602011670号
苏公网安备32010602011670号